

Directions (1-5): Study the following information carefully and answer the questions given below:
Eight persons M, N, O, P, Q, R, S and T are taking ten days leave in eight different months i.e. January, March, April, May, June, July, August and September. Each of them likes different colours i.e. Red, Blue, Green, Pink, Orange, Grey, White and Purple but not necessary in same order.
P takes leave in the month which has even number of days and likes White colour. Three persons take leave between P and R. M takes leave in the month which has odd number of days. Two persons take leave between R and the one who likes red colour. Q takes leave in the month just before O. Equal numbers of person take leave after R and before T, who does not like blue and green colour. M takes leave in the month after the one who likes Orange colour. The one who likes grey colour take leave in August. Same numbers of person take leave after the one who likes orange colour and before the one who likes Blue colour. M does not like blue colour. Q does not take leave in the month which has odd number of days. The person who likes Pink colour take leave before the one who likes Green colour and after the one who likes Purple colour. S does not take leave in the month which have odd number of days.
Q1. Who among the following take leave just before the one who likes Green colour?
(a) The one who likes Pink colour
(b) O
(c) The one who likes Red colour
(d) R
(e) None of these
Q2. N likes which of the following colour?
(a) Blue
(b) Orange
(c) Purple
(d) Green
(e) None of these
Q3. In which of the following month S takes leave?
(a) August
(b) January
(c) September
(d) June
(e) July
Q4. Four of the following five are alike in certain way based from a group, find the one that does not belong to that group?
(a) O
(b) Q
(c) M
(d) T
(e) N
Q5. How many persons take leave between T and R?
(a) One
(b) Five
(c) Three
(d) Four
(e) None
Directions (6-10): Study the information and answer the following questions:
In a certain code language,
“There are numerous ways” is coded as ” X@25 O@21 B#18 U#18 ”
“rightful due political economy” is coded as ” F#13 Q#1 E#21 S@21″
“work culture to provide” is coded as ” X@18 Q#4 D#18 U@20″
Q6.What is the code for ‘requisite’ in the given code language?
(a) R#20
(b) S#18
(c) T#20
(d) S#20
(e)None of these
Q7.What is the code for ‘proximity’ in the given code language?
(a) R#25
(b) M#25
(c) Q#20
(d) L#25
(e)None of these
Q8.What may be the possible code for ‘ visible reminder’ in the given code language?
(a) S#5 W#12
(b) S@5 X#12
(c) S@5 W#12
(d) T@5 W#12
(e)None of these
Q9.What may be the possible code for ‘waiting time’ in the given code language?
(a) X#7 V@5
(b) X@7 U@5
(c) X#7 U#5
(d) X#14 U@13
(e)None of these
Q10.What is the code for ‘publicly enforcing’ in the given code language?
(a) R@25 F#7
(b) Q#25 F#7
(c) Q@25 F@7
(d) Q@12 F#14
(e)None of these
Directions (11-15): Study the given information and answer the questions:
A word and number arrangement machine when given an input line of words and numbers rearranges them following a particular rule in each step. The following is an illustration of an input and rearrangement.
Input: 54 Sea 88 doctor 41 remind 63 aunt united 15 92 Even
Step I: 17 54 Sea 88 doctor 41 remind 63 united 92 Even aunt
Step II: doctor 17 54 Sea 88 remind 63 united 92 Even aunt 43
Step III: 52 doctor 17 Sea 88 remind 63 united 92 aunt 43 Even
Step IV: remind 52 doctor 17 Sea 88 united 92 aunt 43 Even 65
Step V: 86 remind 52 doctor 17 united 92 aunt 43 Even 65 Sea
Step VI: united 86 remind 52 doctor 17 aunt 43 Even 65 Sea 90
And Step VI is the last step of the rearrangement of the above input.
As per the rules followed in the above steps, find out in each of the following questions the appropriate step for the given input.
Input: 48 Stream 82 Damage 35 read 57 apple unit 9 86 end
Q11. In which step the elements ‘11 unit 86’ found in the same order?
(a) Step I
(b) Step II
(c) Step III
(d) Step V
(e)Step VI
Q12. In step IV, which of the following word/number would be at 2nd position to the left of 7th from the right end?
(a) 11
(b) Stream
(c) 82
(d) unit
(e) 86
Q13. How many step remind completing the above arrangement?
(a) Three
(b) Four
(c) Six
(d) Seven
(e) Five
Q14. Which of the following would be the step III after arrangement?
(a) 16 Damage 11 Stream 82 read 57 unit 86 apple 37 end
(b) 6 Damage 11 Stream 80 read 57 unit 86 apple 37 end
(c) 6 Damage 15 Stream 82 read 57 unit 86 apple 37 end
(d) 46 Damage 11 Stream 82 read 57 unit 86 apple 37 end
(e) None of these
Q15. In step VI, ‘80’ is related to ‘unit’ and ‘11’ is related to ‘Damage’. In the same way ‘59’ is related to?
(a) end
(b) unit
(c) read
(d) Stream
(e) None of these
Practice More Questions of Reasoning for Competitive Exams:
Solutions
Solutions (1-5):
Sol.
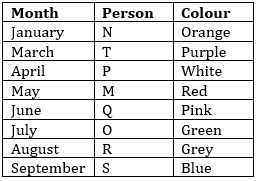
S1.Ans(a)
S2.Ans(b)
S3.Ans(c)
S4.Ans(b)
S5.Ans(d)
Solutions (6-10):
These are the latest pattern of coding-decoding questions. In these questions we are applying following concept:-

S6. Ans.(d)
Sol. S#20
S7. Ans.(c)
Sol. Q#20
S8. Ans.(c)
Sol. S@5 W#12
S9. Ans.(d)
Sol. X#14 U@13
S10. Ans.(d)
Sol. Q@12 F#14
Solution (11-15):
Students let us understand the Logic behind this Question and let’s understand how to solve it. When we see the each step, then we can find that there is both number and words are arranged in each step.
1) For words arrangement- Words are arranged according to alphabetical order given in English dictionary. In first step the words which comes first according to English dictionary arranged first to extreme right. And in second step next word are arranged to extreme left.
And this process is continued in further step.
2) For number arrangement- Number are arranged according to ascending order. In first step lowest number arranged in extreme left. And in second step next number is arranged in extreme right. And this process is continued in further step( Each odd number is added by two(+2) while they are arranged and two is subtracted by each even number(-2) while they are arranged).
Input: 48 Stream 82 Damage 35 read 57 apple unit 9 86 end
Step I: 11 48 Stream 82 Damage 35 read 57 unit 86 end apple
Step II: Damage 11 48 Stream 82 read 57 unit 86 end apple 37
Step III: 46 Damage 11 Stream 82 read 57 unit 86 apple 37 end
Step IV: read 46 Damage 11 Stream 82 unit 86 apple 37 end 59
Step V: 80 read 46 Damage 11 unit 86 apple 37 end 59 Stream
Step VI: unit 80 read 46 Damage 11 apple 37 end 59 Stream 84
S11. Ans.(d)
S12. Ans.(a)
S13. Ans.(c)
S14. Ans.(d)
S15. Ans.(a)
Download PDF of this Reasoning Quiz for IBPS Mains 2020
Practice with Crash Course and Online Test Series for IBPS Mains 2020:
- IBPS RRB PO and Clerk Prime 2020-21 Online Test Series
- IBPS PO Online test series (Prelims + Mains) 2020 by Adda247
- IBPS KA MAHAPACK Online Live Classes
Click Here to Register for Bank Exams 2020 Preparation Material
If you are preparing for IBPS Mains Exam, then you can also check out a video for Reasoning below:
https://www.youtube.com/watch?v=8bxpGNDkMzQ








 Daily Current Affairs and GK Updates (22...
Daily Current Affairs and GK Updates (22...
 IB Security Assistant Interview Call Let...
IB Security Assistant Interview Call Let...
 SBI Strike on 25 & 26 May 2026 - AIS...
SBI Strike on 25 & 26 May 2026 - AIS...


